It’s been almost four years since professors Jennifer Doudna and Emmanuelle Charpentier received the Nobel Prize in Chemistry for discovering the now well-known genome editing CRISPR/Cas9 system. Since then, researchers have tried to utilize this tool to induce targeted alterations in DNA to repair faulty genes or change specific nucleotide sequences in the fight against disease. At the end of 2023, the first CRISPR-based cell therapy was authorized by regulatory agencies against two genetic disorders – sickle cell disease and transfusion-dependent beta thalassemia. This article will discuss the science behind the new treatment and how it can help patients’ everyday lives. But first, let’s look at the aetiology of these two genetic disorders.
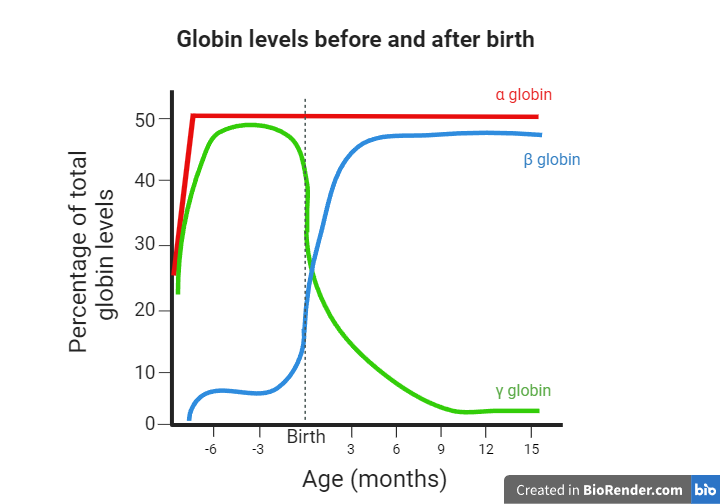
Red blood cells are one of the main components of our blood, and their purpose is to carry oxygen to various tissues in our bodies. This function is mediated by hemoglobin molecules. Hemoglobin (Hb) is a tetrameric protein, meaning that it consists of four polypeptide subunits. Each subunit has an iron-containing heme group that facilitates oxygen delivery to cells. Humans have different types of hemoglobin molecules depending on the polypeptides that form their subunits. For example, HbA is the most abundant hemoglobin in adults, consisting of two α (alpha) and two β (beta) chains. Embryos use a different molecule called fetal hemoglobin (HbF), which consists of two α and two γ (gamma) chains. HbA2 is another type of hemoglobin consisting of two α and two δ (delta) chains, and it is found at a lower percentage. What’s important to remember is that as the third trimester of pregnancy is reached, and even more so after the baby is born, the expression levels of the γ globin-coding gene decline while the β globin-coding gene expression levels rise. As a result, the fetal hemoglobin (HbF) and adult hemoglobin (HbA) levels decline and rise, respectively as well. Consequently, from one year of age and for the rest of their lives, humans use HbA as their main oxygen-transferring molecule, while HbF constitutes about 1% of the total hemoglobin.
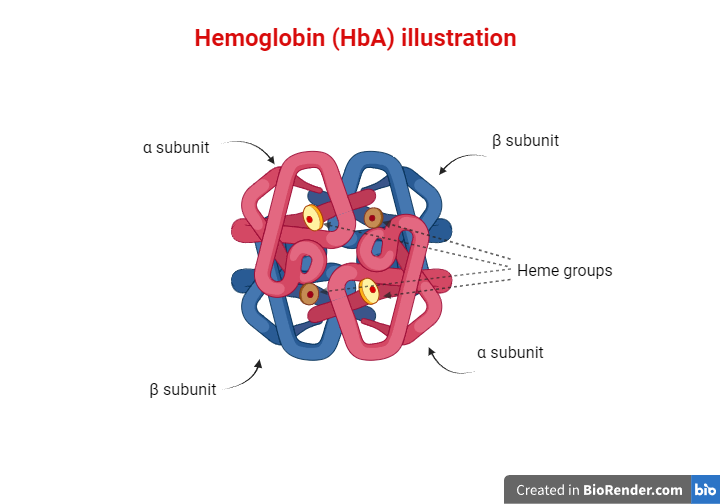
However, in a series of genetic disorders known as hemoglobinopathies, the levels or functions of one or more types of hemoglobin are disrupted due to mutations in the genes that code for their subunits. In this article, we will discuss two of them: sickle cell disease and beta thalassemia.
In sickle cell disease (SCD), the gene coding for the β globin chain presents a single base substitution, which creates the sickle β-globin gene, known as βs. This mutation causes glutamic acid to be replaced by valine at the sixth amino acid position. That is responsible for major alterations in the structure and function of the hemoglobin, leading to a series of symptoms that characterize the sickle cell disease phenotype. Sickle cell disease is an autosomal recessive disease, meaning that people who inherit two copies of the faulty β globin gene suffer from the clinical manifestations of the disorder. That is attributed to the absence of normal β globin in their red blood cells. Consequently, α chains and the abnormal β chains form the sickle tetrameric hemoglobin known as HbS. HbS molecules tend to stick to one another, disrupting the structure of the red blood cells, which become sickle-shaped. As a result, there is no efficient oxygen transport to other tissues. Furthermore, red blood cells can block the blood flow inside blood vessels, leading to several problems that can become life-threatening. Some of the most characteristic clinical manifestations in patients with sickle cell disease include severe anemia, vaso-occlusive episodes, and serious organ damage of the liver, kidneys, or spleen.
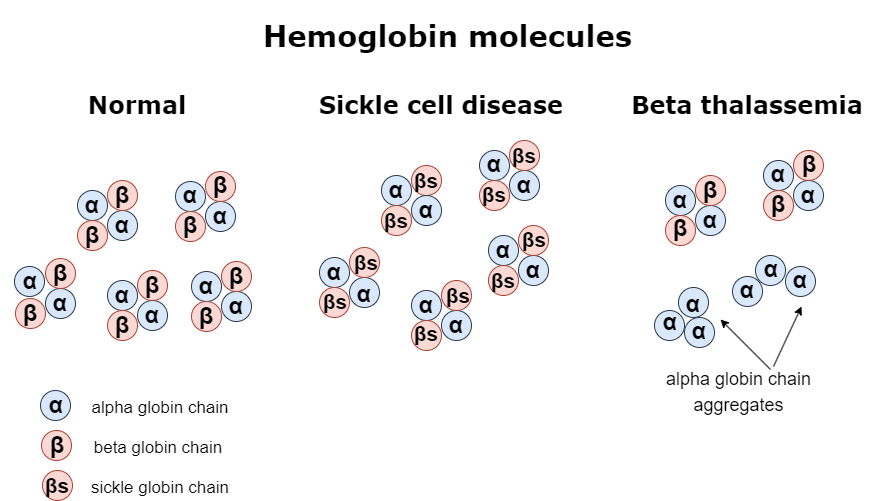
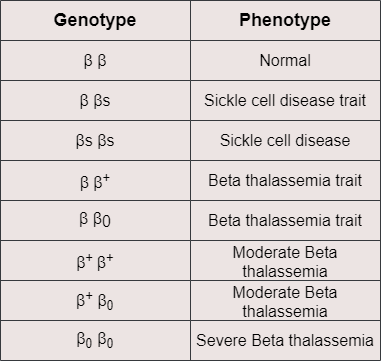
Similarly, beta thalassemia is another genetic disorder that leads to problems related to faulty oxygen transport. Beta thalassemia causes mild to severe anemia depending on the levels of the β globin that is produced due to mutations of the β globin-coding gene. For example, someone might be affected because both copies of the β globin-coding gene carry mutations responsible for reduced production of the β globin. In this case, each affected gene is symbolized as β+. However, it is also possible that due to more serious mutations, the β globin-coding gene, which in this case is symbolized as β0, does not produce any β globin polypeptide. That means different combinations of inherited β globin genes, as shown in the table below, can have a variable effect on the total production of the hemoglobin. More particularly, the less β globin is produced, the more severe the disease is. Moreover, when there is not enough β globin to form HbA tetrameric proteins, the α chains that are in excess tend to bind to each other and create α globin aggregates that negatively affect erythrocyte functioning and cause intravascular hemolysis. For the above reasons, patients with beta thalassemia suffer from anemia, which needs to be addressed with frequent blood transfusions. They might also present bone abnormalities, jaundice, and liver or spleen enlargement.
Patients who suffer from sickle cell disease or beta thalassemia are subjected to symptomatic management in order to alleviate pain or prevent serious complications. Anemia and its subsequent side effects are often treated with frequent blood transfusions. However, iron overload caused by the large amounts of transfused red blood cells may lead to intoxication and organ damage, and this is something that needs to be addressed with medicines. So far, bone marrow or stem cell transplantation has been the only effective cure for both sickle cell disease and beta thalassemia, provided that there is a suitable donor. After transplantation, the body produces healthy red blood cells that replace the defective ones. Of course, one should always examine whether the potential benefits outweigh the possible risks of such a treatment before a patient is subjected to it. Therefore, it is clear there is a need to develop new, effective therapies against these disorders.
As was mentioned earlier, the γ globin gene expression levels start to decline before birth, and shortly afterward, the fetal hemoglobin (HbF) reaches about 1% of the total hemoglobin in the human body. However, one interesting finding is that some people produce higher than average amounts of HbF even after the first year of their life. This phenomenon, called Hereditary Persistence of Fetal Hemoglobin (HPFH), has been attributed to variations in genomic regions that control the γ globin-coding gene expression. Also, what’s more important is that individuals with sickle cell disease or beta thalassemia that present HPFH can manage their disease better. They are less prone to disease-associated symptoms, less likely to be hospitalized, and don’t require frequent blood transfusions. Consequently, the study of the mechanisms underlying the persistence of fetal hemoglobin could offer valuable information for treating these genetic disorders.
Molecular studies have unraveled the implication of a molecule named BCL11A in the hemoglobin switch mechanism. BCL11A is a transcription factor that represses the expression of the γ globin gene and leads to a reduction in fetal hemoglobin levels. At the same time, it enhances the β globin gene transcription, causing an increase in adult hemoglobin levels, respectively. So, if scientists could somehow prevent BCL11A from repressing the production of the γ globin, patients’ health could be improved.
It seems now that research in this field has come to fruition. A CRISPR-based gene editing cell therapy that targets BCL11A has been developed, offering new hope to patients suffering from these two genetic disorders. Since December 2023, Casgevy or exagamglogene autotemcel (exa-cel), produced by Vertex Pharmaceuticals, is the first gene editing treatment that has been authorized by both the FDA (in the US) and the EMA (in Europe) for use in patients with sickle cell disease or transfusion-dependent beta thalassemia.
CRISPR/Cas9 is a new technology that uses molecular scissors to specifically target regions of interest in the genome in order to replace, add, or remove sequences for research, preventive, or treatment purposes. CRISPR/Cas is a bacterial immune system used for the protection against foreign genetic material, such as bacteriophages. For example, when a bacterial cell encounters a foreign DNA molecule for the first time, part of this genetic material is integrated as a spacer in an array of sequences, the CRISPR array. This way, the cell can acquire memory against this particular invader. If that cell encounters the same invader again, the spacer sequence is transcribed to RNA. The RNA molecule then recognizes the complementary sequence of the foreign material, and a Cas endonuclease cleaves the foreign molecule, thus protecting the host from infection. Professors Jennifer Doudna and Emmanuelle Charpentier, who discovered this defensive mechanism, were awarded the Nobel Prize in Chemistry in 2020. It was instantly clear that their CRISPR/Cas discovery could become a very promising tool in the fields of genome editing and gene therapy.
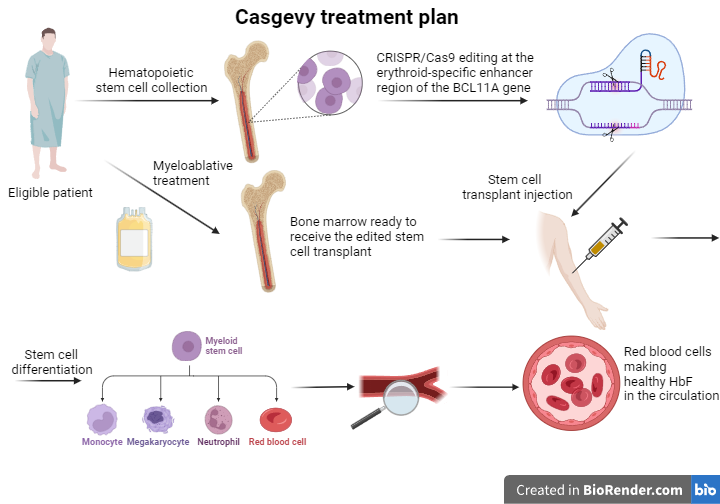
In the case of Casgevy, the CRISPR/Cas system uses a target-specific sequence to guide the Cas endonuclease to the erythroid-specific enhancer region of the BCL11A gene of hematopoietic stem cells. A double-stranded DNA break is then created, allowing scientists to cause alterations in the nucleotide sequence. This DNA editing process leads to a reduction in the expression of the BCL11A gene, thus disrupting the mechanism that normally drives the switch from fetal to adult hemoglobin.
Patients eligible for this new treatment are administered a drug that enables stem cell mobilization from the bone marrow. This way, these cells can then be collected from the bloodstream. Later, these stem cells are processed accordingly, and with the CRISPR/Cas9 technology, their genome is edited at the erythroid-specific enhancer region of the BCL11A gene. In order for the treatment to be effective, patients must first undergo a myeloablative conditioning therapy that kills the defective cells and makes room for the engraftment of the in vitro edited stem cells. After the successful completion of the previous steps, Casgevy, in the form of patient-specific edited hematopoietic stem cells, is administered to the patient intravenously. The edited stem cells engraft in the bone marrow and start multiplying before differentiating to the various types of blood cells. If everything goes accordingly, the erythroid precursor cells that are produced will present a reduction in the BCL11A gene transcription and will be able to synthesize higher levels of the γ globin chain than before. Therefore, the red blood cells that will end up in the patient’s circulation will have elevated levels of the HbF hemoglobin. The presence of the fetal hemoglobin will compensate for the faulty β globin synthesis, and the patient’s overall health will improve.
Casgevy is now available in many countries for patients suffering from sickle cell disease with frequent vaso-occlusive episodes or transfusion-dependent beta thalassemia that are 12 years of age or older. Of course, as with every treatment, there is a possibility that patients may experience side effects. Since the edited stem cells require some time to engraft, the body will not have enough white blood cells or platelets, putting them at risk of infections or bleeding, respectively. Moreover, due to the use of a myeloablative conditioning regimen, patients must use contraception for six months or stop breastfeeding. Since fertility can also be affected by myeloablation, patients may want to discuss gamete preservation options. One of the main concerns of scientists about using CRISPR for genome editing has been the risk of off-target effects, which means editing DNA at sites where they do not wish to change the nucleotide sequence. In the case of Casgevy, studies have not shown any unwanted DNA edits, but researchers continue their efforts to find ways to prevent this from happening in general.
As far as the effectiveness of Casgevy is concerned, clinical trials have shown that patients who have received the treatment have not rejected their edited stem cell transplant. It was also observed that over 90% of patients with sickle cell disease who were treated with Casgevy have avoided hospitalizations due to severe complications, and they haven’t experienced any severe vaso-occlusive episodes in the 12 months since receiving the treatment. In the group of patients suffering from transfusion-dependent beta thalassemia, patients did not require transfusions for at least a year following the Casgevy treatment at a similar percentage. As Casgevy becomes available in specialized treatment centers in different countries, it is expected that follow-up studies that will last years after treatment will offer more information regarding its benefits.
Sources
https://pubmed.ncbi.nlm.nih.gov/33283989/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3092400/pdf/nihms287521.pdf